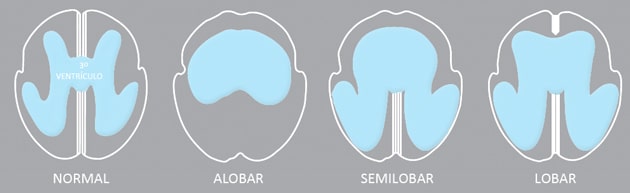
Sinal do Polvo
Em 2009 Rabih Chaoui e colaboradores descreveram a translucência intracraniana como método para rastrear espinha bífida. A translucência intracraniana é a aparência ultrassonográfica do quarto ventrículo no plano sagital médio, como visto no momento do exame de 11 a 14 semanas.
O valor médio da translucência intracraniana está entre 1,5 mm a 2,5 mm para o comprimento cabeça-nádegas (CCN) de 45 mm a 84 mm, respectivamente.
Em 2013 Elena Andreeva descreveu no site TheFetus.Net um novo “sinal de polvo” (Octopus-like sign) para diagnóstico de espinha bífida e malformação de Dandy Walker durante o exame ultrassonográfico de primeiro trimestre entre 11 e 14 semanas de gestação.
O princípio do método é obter no mesmo corte da translucência nucal imagem que demonstre o tálamo, tronco cerebral, mesencéfalo, quarto ventrículo e cisterna magna. O arranjo anatômico dessas estruturas no plano sagital mediano lembra um pequeno polvo.
Aparência normal do Sinal do Polvo
A aparência normal do sinal do polvo deve ter as seguintes características:
- Ambos os braços do polvo têm o mesmo tamanho;
- O plexo coróide deve ser visto dentro do quarto ventrículo e o segundo braço do “polvo” que compreende o mesencéfalo e o quarto ventrículo deve ser claramente separado do primeiro braço do “polvo” (tronco cerebral).
- A cisterna magna deve ser claramente separada do segundo braço do “polvo” (mesencéfalo e quarto ventrículo).
Alterações que podem ser identificadas
As alterações que podem ser identificadas incluem especialmente as alterações da fossa posterior como a mielomeningocele e a malformação de Dandy-Walker. Veremos abaixo quais as alterações esperadas nestas malformações.
Mielomeningocele
Nos casos de mielomeningocele a imagem do polvo habitualmente é alterada da seguinte forma:
- Aumenta o tamanho do primeiro braço do “polvo” (tronco cerebral) ;
- Diminuição do tamanho do segundo braço do “polvo” (apenas o plexo coróide dentro do quarto ventrículo muito pequeno está presente);
- A cisterna magna não é visível.
Malformação de Dandy-Walker
Já na malformação de Dandy-Walker as alterações ultrassonograficas são diferentes. A aplasia do vermis cerebelar leva ao alargamento do segundo braço do “polvo” devido à fusão do quarto ventrículo, cisterna magna e plexo coróide.
A importância deste sinal ultrassonográfico
O sinal do polvo é uma maneira subjetiva e simples de avaliar a translucência intracraniana. A translucência intracraniana é a aparência ultrassonográfica do quarto ventrículo no plano sagital médio, utilizado no exame entre 11 e 14 semanas para a medida da translucência nucal. É usado como um marcador para defeitos do tubo neural. Sua ausência é relatada na espinha bífida aberta.
O diagnóstico precoce desta anomalia é importante pois hoje sabemos que o diagnóstico precoce da mielomeningocele permite a realização da cirurgia fetal para correção da mielomeningocele. Esta conduta permite uma melhora no prognóstico para as crianças afetadas por esta malformação.
A cirurgia fetal para mielomeningocele reduz significativamente a necessidade de derivação ventrículo-peritonial para redução da ventriculomegalia. Também sabemos pelo estudo MOMs que a chance da criança conseguir deambular é significativamente maior quando a correção da mielomeningocele é feita intra-útero.
Enfim, o mesmo plano utilizado para medir a translucência nucal pode ser utilizada para identificar precocemente malformações do sistema nervoso central.