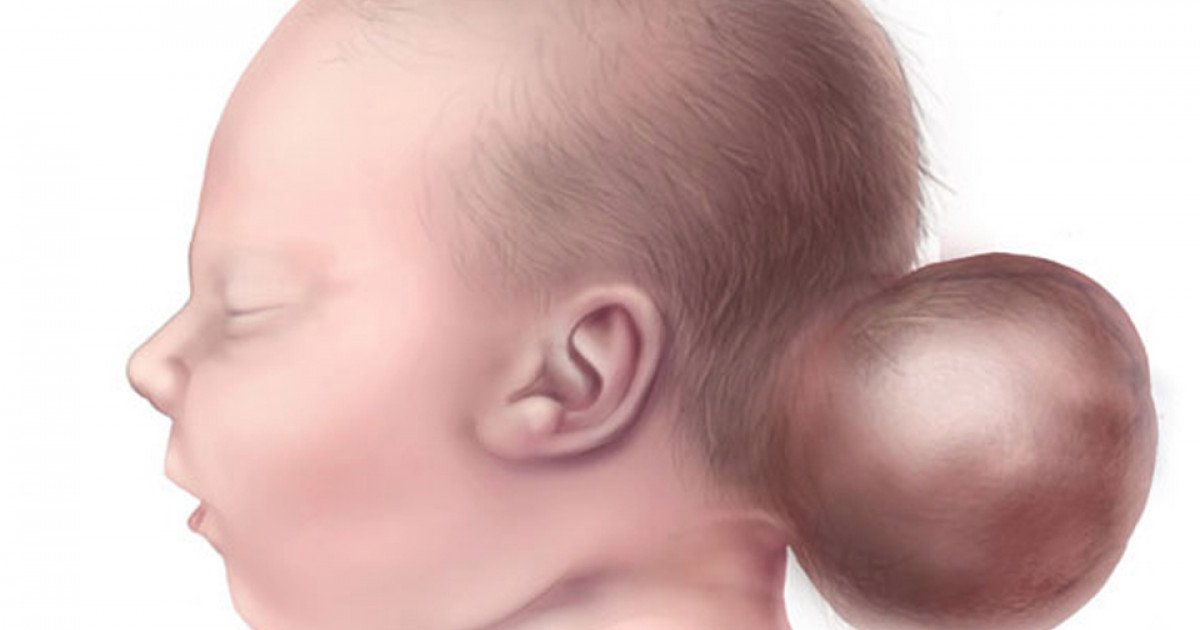
Onfalocele é uma herniado das vísceras abdominais através de um defeito central do abdômen, no local de inserção do cordão umbilical. As vísceras herniadas são compostas por 3 camadas: peritônio, âmnio e geléia de Wharton. A onfalocele difere da gastrosquise por ser coberta por membrana que recobre o conteúdo abdominal e por ser mais frequentemente associada com alterações cromossômicas.
Como a onfalocele se forma: embriologia em linguagem acessível
Para entender a onfalocele, é útil conhecer um processo normal do desenvolvimento fetal. Por volta da 6ª semana de gestação, o intestino médio cresce rapidamente e, temporariamente, se projeta para fora da cavidade abdominal — dentro do próprio cordão umbilical. Isso é completamente fisiológico. Na 10ª semana, a cavidade abdominal já é suficientemente grande para receber esses órgãos de volta, e o retorno acontece normalmente.
Na onfalocele, esse retorno não ocorre de forma completa. Os órgãos abdominais — intestino, fígado, estômago — permanecem herniados através do defeito na parede abdominal e ficam contidos dentro de um saco membranoso composto por peritônio e âmnio. Essa membrana é a principal característica que distingue a onfalocele de outros defeitos da parede abdominal.
Onfalocele ao ultrassom: o que o exame mostra
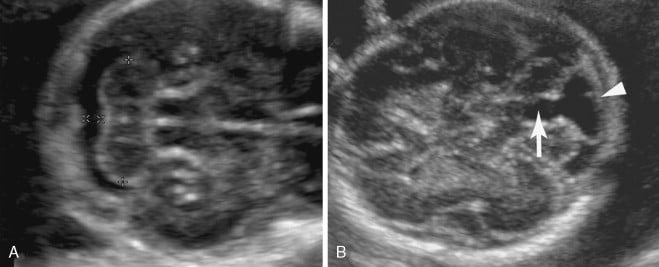
A onfalocele é identificada ao ultrassom como uma massa heterogênea na linha média do abdômen fetal, na base de inserção do cordão umbilical. O diagnóstico pré-natal é possível em mais de 90% dos casos, sendo que:
- Onfaloceles contendo apenas intestino podem ser diagnosticadas de forma confiável após a 12ª semana — antes disso, é difícil diferenciá-las da hérnia fisiológica normal do intestino médio
- Onfaloceles contendo fígado podem ser detectadas já entre 9 e 10 semanas, pois a presença do fígado no saco é sempre um achado patológico
- A membrana intacta ao redor do conteúdo herniado é o sinal ultrassonográfico mais característico
- O cordão umbilical se insere diretamente no ápice do saco onfalocélico
O tamanho da onfalocele varia amplamente. Aproximadamente 80% das onfaloceles contêm parte do fígado. As chamadas “onfaloceles gigantes” — que contêm a maior parte do fígado ou medem mais de 5 cm — representam um subgrupo com desafios cirúrgicos e prognóstico específicos.
Onfalocele × Gastrosquise: como diferenciar ao ultrassom
Essa é uma das distinções mais importantes na medicina fetal, pois as duas condições têm causas, associações e condutas muito diferentes:
| Característica |
Onfalocele |
Gastrosquise |
| Localização do defeito |
Linha média — base do cordão umbilical |
Paraumbilical, geralmente à direita |
| Membrana cobrindo vísceras |
Presente (peritônio + âmnio) |
Ausente — órgãos expostos ao líquido amniótico |
| Inserção do cordão |
No ápice do saco |
Normal, lateral ao defeito |
| Fígado herniado |
Possível (em ~80%) |
Raro |
| Associação com aneuploidias |
Alta (30–50% dos casos) |
Baixa |
| Associação com síndromes genéticas |
Alta (Beckwith-Wiedemann, Pentalogy of Cantrell) |
Muito baixa |
Associação com cromossomopatias e síndromes genéticas
A onfalocele tem associação significativa com anomalias cromossômicas — essa é uma das razões pelas quais o diagnóstico pré-natal tem tanto impacto no planejamento clínico. Os dados mais relevantes:
- Em onfaloceles diagnosticadas entre 11 e 13 semanas com conteúdo exclusivamente intestinal, a taxa de defeitos cromossômicos pode chegar a 30–50%, predominando a Trissomia 18 (Síndrome de Edwards) e a Trissomia 13 (Síndrome de Patau)
- Vale notar que onfaloceles intestinais detectadas no primeiro trimestre têm resolução espontânea em até 90% dos casos — é o retorno tardio do intestino ao abdômen
- Onfaloceles contendo fígado têm menor taxa de aneuploidia, mas maior associação com a Síndrome de Beckwith-Wiedemann — condição genética de hipercrescimento fetal presente em cerca de 10% dos casos
- Anomalias estruturais associadas são encontradas em 35 a 70% dos casos, incluindo cardiopatias congênitas, anomalias genitourinárias, defeitos do tubo neural e malformações gastrointestinais
Por isso, o diagnóstico de onfalocele ao ultrassom deve sempre motivar uma investigação complementar estruturada: avaliação morfológica fetal completa, ecocardiograma fetal e discussão sobre cariótipo fetal (amniocentese ou biópsia de vilo corial).
Conduta e acompanhamento durante a gestação
Após o diagnóstico, o acompanhamento é realizado por uma equipe multidisciplinar — especialista em medicina fetal, neonatologista e cirurgião pediátrico — e inclui:
- Avaliação morfológica detalhada para identificar malformações associadas
- Ecocardiograma fetal — cardiopatias estão entre as anomalias mais frequentemente associadas
- Investigação genética — amniocentese ou biópsia de vilo corial para cariótipo e, conforme indicado, pesquisa de Síndrome de Beckwith-Wiedemann
- Ultrassonografias seriadas a cada 3–4 semanas para monitorar o crescimento fetal e o volume de líquido amniótico
- Vigilância fetal intensificada a partir de 32 semanas (cardiotocografia ou perfil biofísico semanal), pois fetos com onfalocele têm maior risco de morte fetal tardia
O planejamento do parto deve incluir a transferência para um centro terciário com UTI neonatal e bloco cirúrgico pediátrico. O parto prematuro eletivo não traz benefícios para o recém-nascido e está associado a maior morbimortalidade. A via de parto segue critérios obstétricos habituais; a cesariana é reservada para onfaloceles gigantes com risco de ruptura de membrana ou distocia.
Tratamento após o nascimento
O tratamento da onfalocele é cirúrgico, realizado pela equipe de cirurgia pediátrica. A abordagem depende principalmente do tamanho do defeito e da presença de outras condições associadas:
- Fechamento primário: indicado para onfaloceles pequenas (defeito menor que 2 cm), realizável nas primeiras horas após o nascimento
- Fechamento em etapas com silo: para onfaloceles maiores, um envoltório artificial protege o conteúdo herniado enquanto ele é gradualmente reduzido para a cavidade abdominal ao longo de dias a semanas
- Fechamento após epitelização: em casos selecionados de onfaloceles gigantes, a membrana é tratada topicamente até que ocorra epitelização espontânea, com reparo cirúrgico definitivo em um segundo tempo
Além da cirurgia, os cuidados neonatais incluem suporte respiratório, nutrição parenteral, controle térmico e prevenção de infecções. O prognóstico é determinado principalmente pela presença e gravidade das anomalias associadas — não pela onfalocele em si.
Perguntas frequentes sobre onfalocele
A onfalocele tem cura?
Sim. A onfalocele tem tratamento cirúrgico estabelecido. O prognóstico depende principalmente do tamanho do defeito e da presença de outras malformações ou cromossomopatias associadas. Onfaloceles isoladas, sem anomalias associadas, têm boas perspectivas com cirurgia pediátrica especializada.
Qual a diferença entre onfalocele e gastrosquise?
As duas são defeitos da parede abdominal fetal, mas diferem em localização, anatomia e associações. Na onfalocele, o defeito é na linha média (base do cordão umbilical) e os órgãos são cobertos por uma membrana. Na gastrosquise, o defeito é lateral ao cordão, os órgãos ficam expostos ao líquido amniótico sem membrana protetora, e a associação com cromossomopatias é muito menor.
A onfalocele sempre indica problema cromossômico?
Não necessariamente. A associação com cromossomopatias existe e é significativa — especialmente com Trissomia 18 e Trissomia 13 — mas não é universal. Onfaloceles contendo fígado têm menor risco de aneuploidia e maior associação com síndromes genéticas como Beckwith-Wiedemann. A investigação genética é sempre recomendada, mas o resultado pode ser completamente normal.
Em que semana é possível diagnosticar a onfalocele ao ultrassom?
Onfaloceles contendo fígado podem ser detectadas já a partir de 9 a 10 semanas. Onfaloceles apenas com intestino são diagnosticadas de forma confiável após a 12ª semana — antes disso, podem ser confundidas com a hérnia intestinal fisiológica, que é um processo normal do desenvolvimento fetal.
O bebê com onfalocele vai precisar de cirurgia ao nascer?
Sim, o tratamento é cirúrgico em todos os casos. O momento e a técnica dependem do tamanho da onfalocele e das condições gerais do recém-nascido. Onfaloceles pequenas podem ser fechadas nas primeiras horas de vida; onfaloceles maiores podem exigir abordagem em etapas ao longo de dias ou semanas.